PME-ONIOM法[1]
諸熊らはONIOM (our Own N-layered integrated molecular Orbital and molecular Mechanics)法[2]を開発し QM計算と分子力場(MM)計算を適切に結びつけることに成功しました。しかし、その適用範囲は有限系もしくは 連続誘電体モデルを用いた溶液までであり、溶媒の全域にわたる明示的な構造は考慮されませんでした。
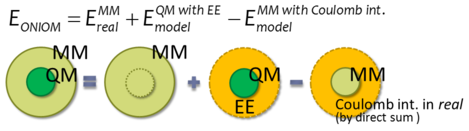
図1.有限系でのONIOMモデル
一方、古典MDシミュレーションの分野では、溶媒分子は周期境界条件により明示的に取り扱われ、 特に静電相互作用の計算では particle-mesh Ewald 和(PME)法[3]が広く用いられてきました。
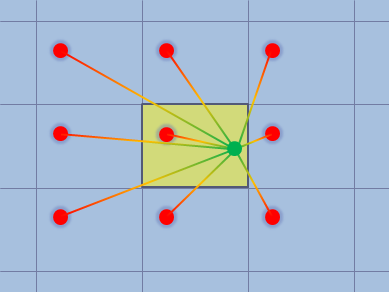
図2.Particle-mesh Ewald和法
当研究室で新規に開発したPME-ONIOM法はONIOM法の外側にPME法による電場を配置し、溶液の明示的な構造を計算に取り込みつつ、 溶質に関してはONIOM法を用いた量子化学計算を可能にする手法です。その概念図を以下に示します。
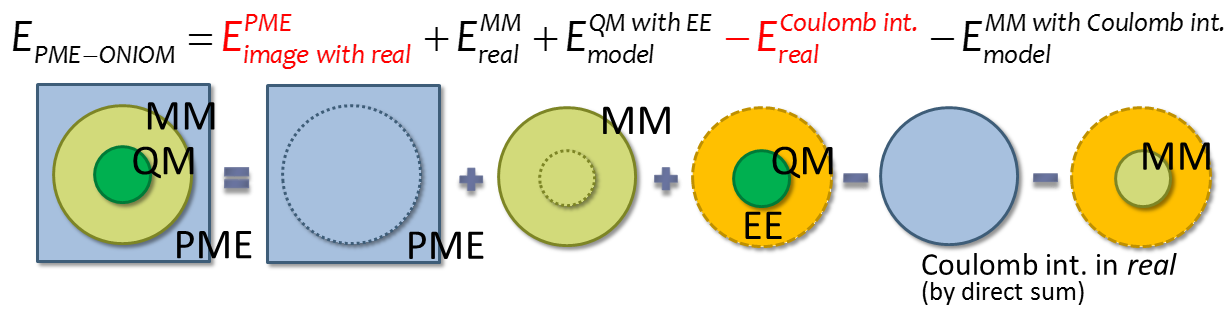
図3.PME-ONIOMモデル
PME-ONIOMモデルはPME法の正確さと量子化学計算の汎用性を両立しており、溶液内での化学反応の高精度な計算を可能にします。
PME-ONIOM法の非断熱遷移への応用[1]
(Z)-penta-2,4-dieniminium cation (PSB3)はロドプシンの発色団のモデル分子(図2)であり、 非断熱遷移を伴う2重結合の異性化が溶液内で遅くなることが実験的に知られていました[4,5]。 しかしその傾向は理論計算の観点からは正しく再現されず25年もの間様々な議論が展開されてきました。 我々はPME-ONIOM法を朱-中村trajectory surface hopping (ZN-TSH) 法[6]とともに用い、 メタノール中のPSB3の非断熱分子動力学シミュレーション非断熱分子動力学シミュレーションに 応用したすることで、溶液内での異性化が遅くなることを再現することに成功いたしました。
また、比較のためにPME法の代わりにMinimum-image conventionを用いたシミュレーションも行い 結果をPME-ONIOM法と比較したところ、Minimum-image conventionではPME法ほどの異性化の遅れは見られず、 その結果がPME法により溶媒を扱った結果であることを確認しました。これにより、
- 溶質の遠くにある溶媒分子も異性化が遅くなる原因となっていること
- PME-ONIOM法が遠距離の相互作用も正しく評価していること
を示しています。
- [1] O. Kobayashi and S. Nanbu, Chem. Phys., 461, 47-57 (2015).
- [2] L. W. Chung, H. Hirao, X. Li, K. Morokuma, WIREs Comput. Mol. Sci. 00, 1-24 (2011).
- [3] T. Darden, D. York, L. Pedersen, J. Chem. Phys., 98, 10089-10092 (1993).
- [4] H. Kandori, H. Sasabe, Chem. Phys. Lett., 216, 126-172 (1993).
- [5] S. L. Logunov, L. Song, M. A. El-Sayed, J. Phys. Chem., 100, 18586-18591 (1996).
- [6] H. Nakamura, Nonadiabatic Transitions: Concepts, Basic Theories and Applications, 2nd Edition, World Scientific, Singapore, 2012.